- 移动端


上海百趣生物科技有限公司
13 年
手机商铺
- NaN
- 0.7000000000000002
- 0.7000000000000002
- 2.7
- 2.7
推荐产品





公司新闻/正文
Circulation(IF=38.7)|南科大医学院覃刚健团队重大发现:靶向Sam68解锁心肌葡萄糖氧化,开启心衰代谢干预新策略
0 人阅读发布时间:2026-06-24 09:47

标题: Sam68 Exacerbates Pathologic Cardiac Hypertrophy by Suppressing Cardiomyocyte Glucose Oxidation
发表期刊: Circulation
影响因子: 38.7
研究背景
心力衰竭是多种心血管疾病的终末阶段,而心肌能量代谢的重编程是其核心病理特征之一。在病理性心肌肥厚过程中,心肌细胞逐渐丧失"代谢灵活性":脂肪酸氧化能力下降,糖酵解虽然代偿性增强,却与线粒体中的葡萄糖氧化发生"解偶联",导致ATP生成速率下降,最终推动心功能持续恶化。然而,连接应激信号与葡萄糖氧化抑制的关键上游调控机制,长期以来尚未得到清晰地阐明。Sam68是压力超负荷诱导心肌代谢重塑的关键驱动因子,其通过"Sam68-Src-STAT3-PDK4"信号轴抑制丙酮酸脱氢酶(PDH)活性、阻断葡萄糖氧化,加速病理性心肌肥厚及心衰进展。遗传敲除或药理学靶向该通路可显著恢复心肌能量代谢、改善心脏重构,为代谢性心力衰竭的精准干预提供了全新可成药靶点。
研究概述
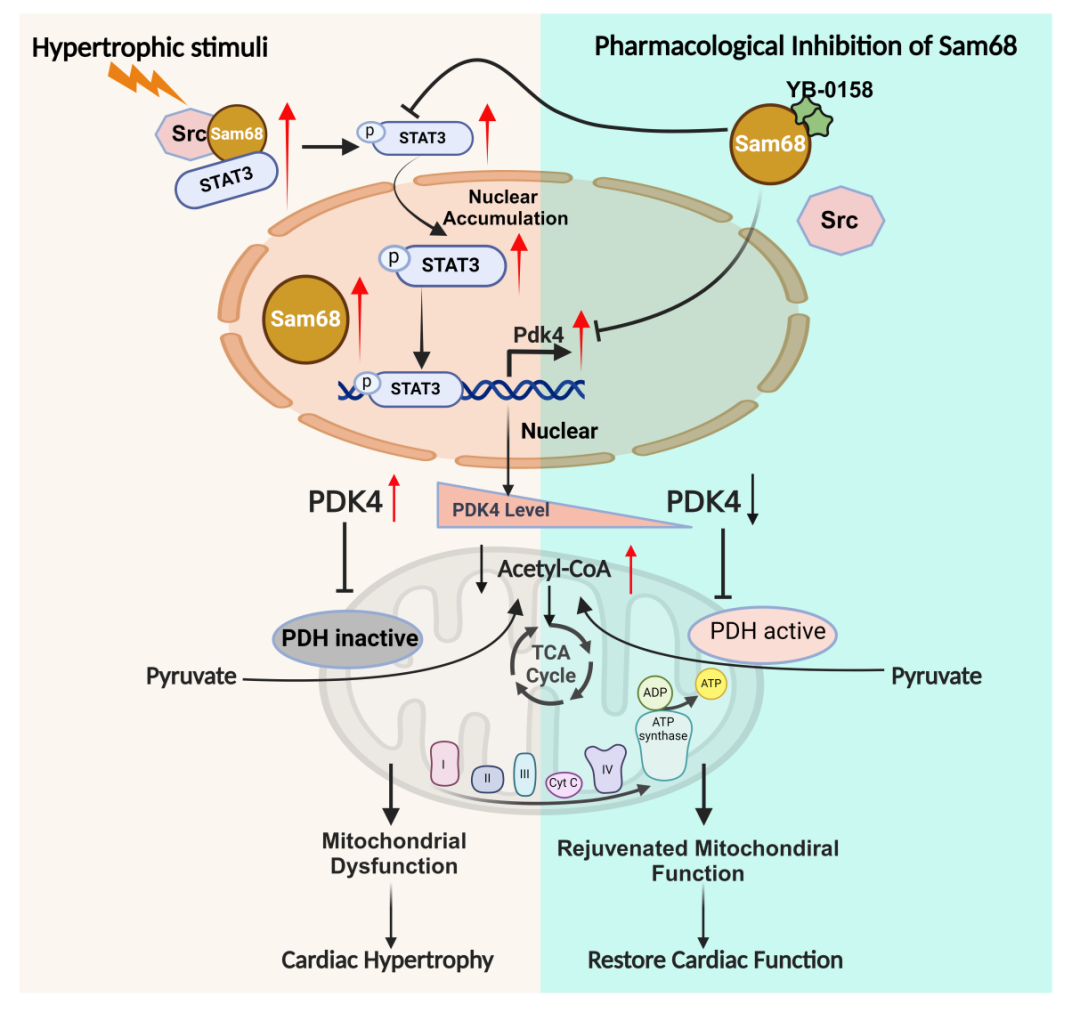
研究结果
01.Sam68在衰竭心脏的心肌细胞中显著上调
为阐明Sam68在心脏疾病中的表达特征,研究团队首先分析了多个独立的左心室转录组数据集,涵盖非衰竭心脏及扩张型、缺血性和肥厚型心肌病样本。结果显示,编码Sam68的KHDRBS1转录本在所有疾病队列中均显著上调。单细胞RNA测序数据集进一步发现,Sam68主要富集于衰竭心肌细胞中。在血管紧张素II(Ang II)输注和主动脉缩窄(TAC)小鼠模型中,上述发现得到了进一步验证:压力应激可迅速诱导Sam68在心肌细胞核内积聚(图1)。
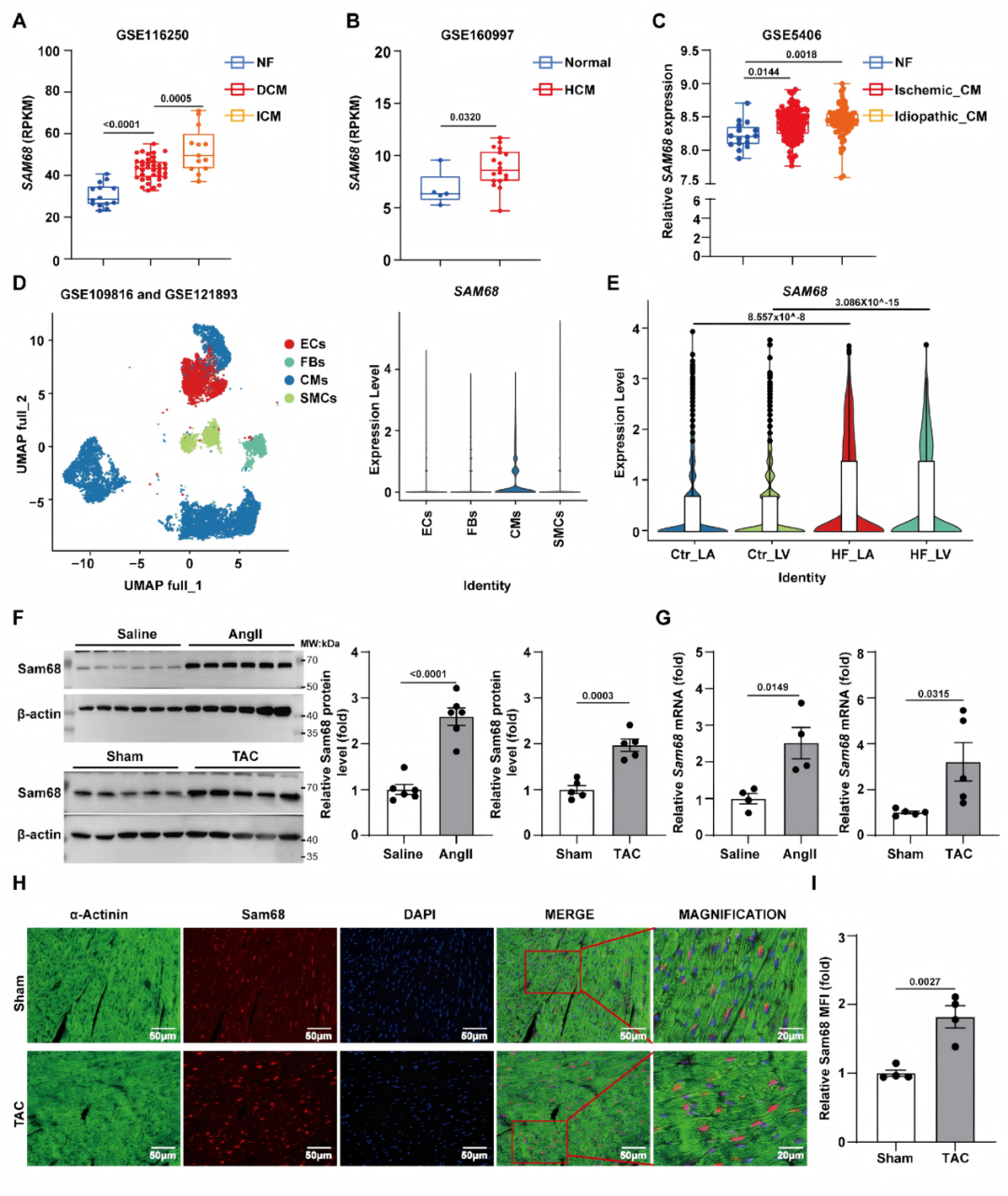
图1.Sam68在人类衰竭心脏及小鼠压力超负荷肥厚模型中表达上调
02.心肌细胞Sam68缺失保护TAC诱导的病理性心肌肥厚
为明确Sam68在压力超负荷诱导的心肌肥厚中的作用,研究团队构建了诱导性心肌细胞Sam68敲除小鼠(Sam68cKO)及同窝对照(CTR)。发现心肌细胞特异性敲除Sam68能改善TAC诱导的病理性心肌肥厚的表型,表现为左心室容积减少,心脏收缩功能改善,心重指数、心肌细胞横截面积及间质纤维化均降低,同时抑制心肌肥厚相关指标(ANP和BNP)的增加(图2)。
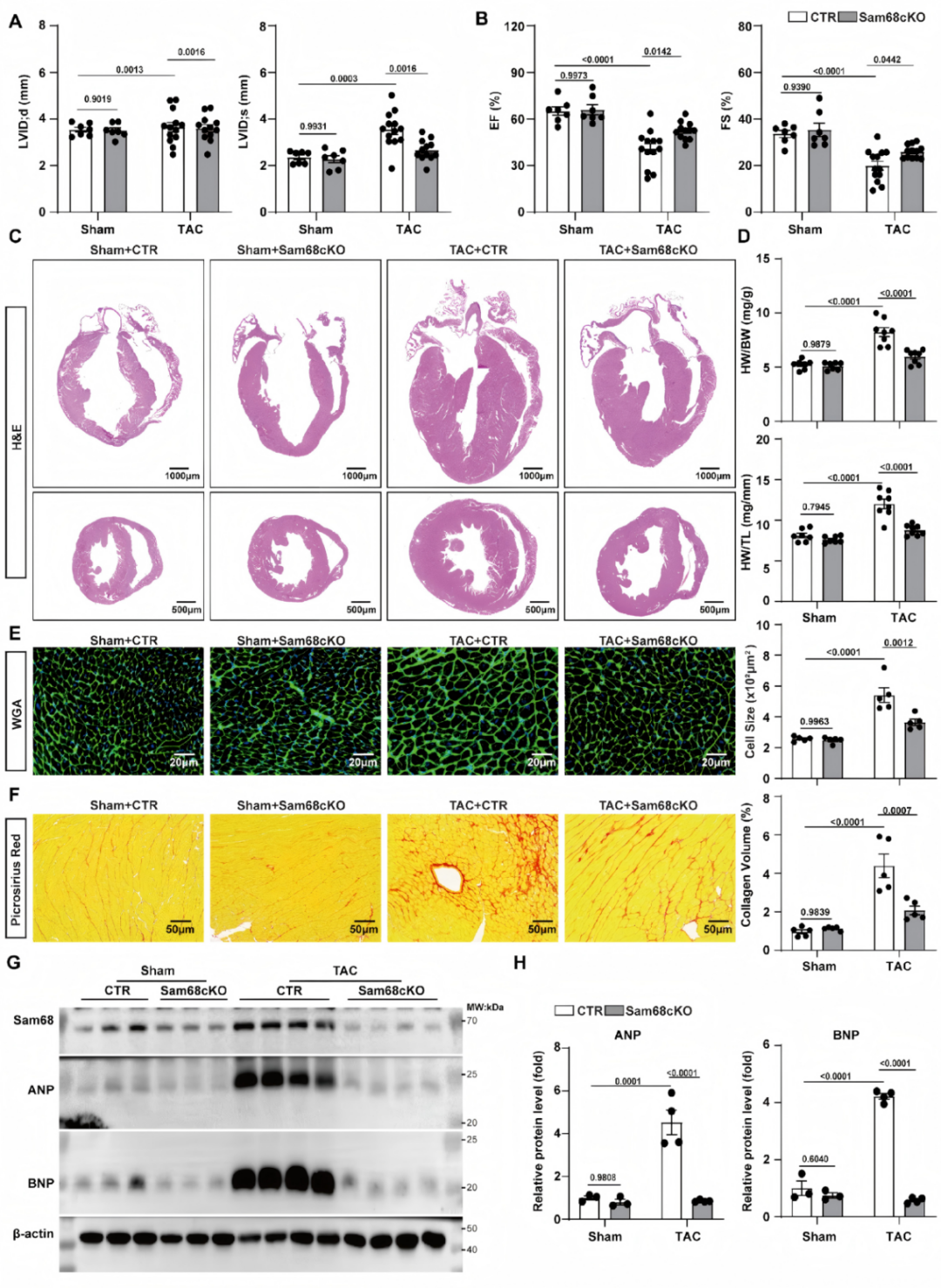
图2.心肌细胞特异性Sam68敲除改善TAC诱导的心脏重构与功能障碍
03.心肌细胞特异性Sam68过表达加剧心肌肥厚与功能障碍
相反,通过递送系统在小鼠心肌细胞特异性过表达Sam68,可进一步加重TAC诱导的心肌肥厚和心脏收缩功能障碍,伴有心肌细胞横截面积增大、心脏纤维化持续加重及ANP和BNP进一步升高。表明Sam68表达升高足以加重TAC诱导的病理性心肌重构(图3)。
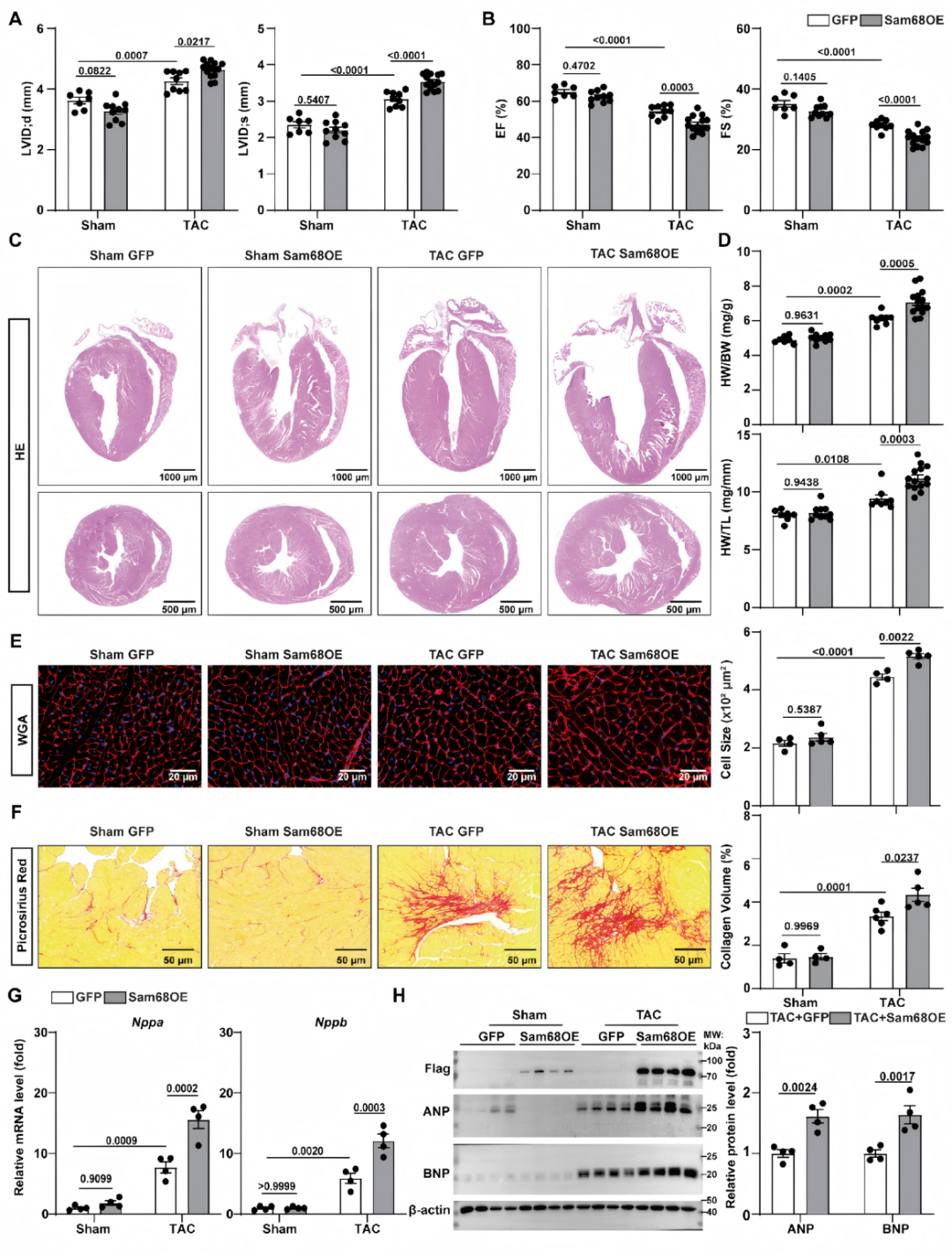
图3.心肌细胞Sam68过表达加重病理性心肌肥厚
04.Sam68缺失保护压力超负荷下的心肌细胞葡萄糖氧化代谢
RNA-seq显示,心肌细胞Sam68缺失抑制肥厚相关基因程序并富集氧化磷酸化通路,提示其维持了线粒体氧化代谢能力。靶向代谢组学分析进一步证实,在压力超负荷早期,Sam68敲除心脏生酮反应减弱、三羧酸循环中间产物得以保留;至慢性阶段则重塑底物利用,表现为上游糖代谢通路中间产物增多,同时AMP和ADP水平降低,能量应激状态得以缓解。体内[U-13C]葡萄糖代谢流实验直接证明,Sam68缺失可恢复TCA循环中间产物的13C标记,增强丙酮酸脱氢酶依赖的氧化途径,并减轻慢性糖酵解过度活化,从而纠正压力超负荷状态下糖酵解与葡萄糖氧化的解偶联。进一步的转录和蛋白分析显示,Sam68缺失使PDH抑制性激酶Pdk4的转录及蛋白水平显著降低,伴随PDH磷酸化下降和活性恢复;相反,Sam68过表达则加剧TAC诱导的PDK4和p-PDHα上调,进一步损伤PDH活性,表明Sam68是压力超负荷下激活PDK4所必需的上游因子(图4)。
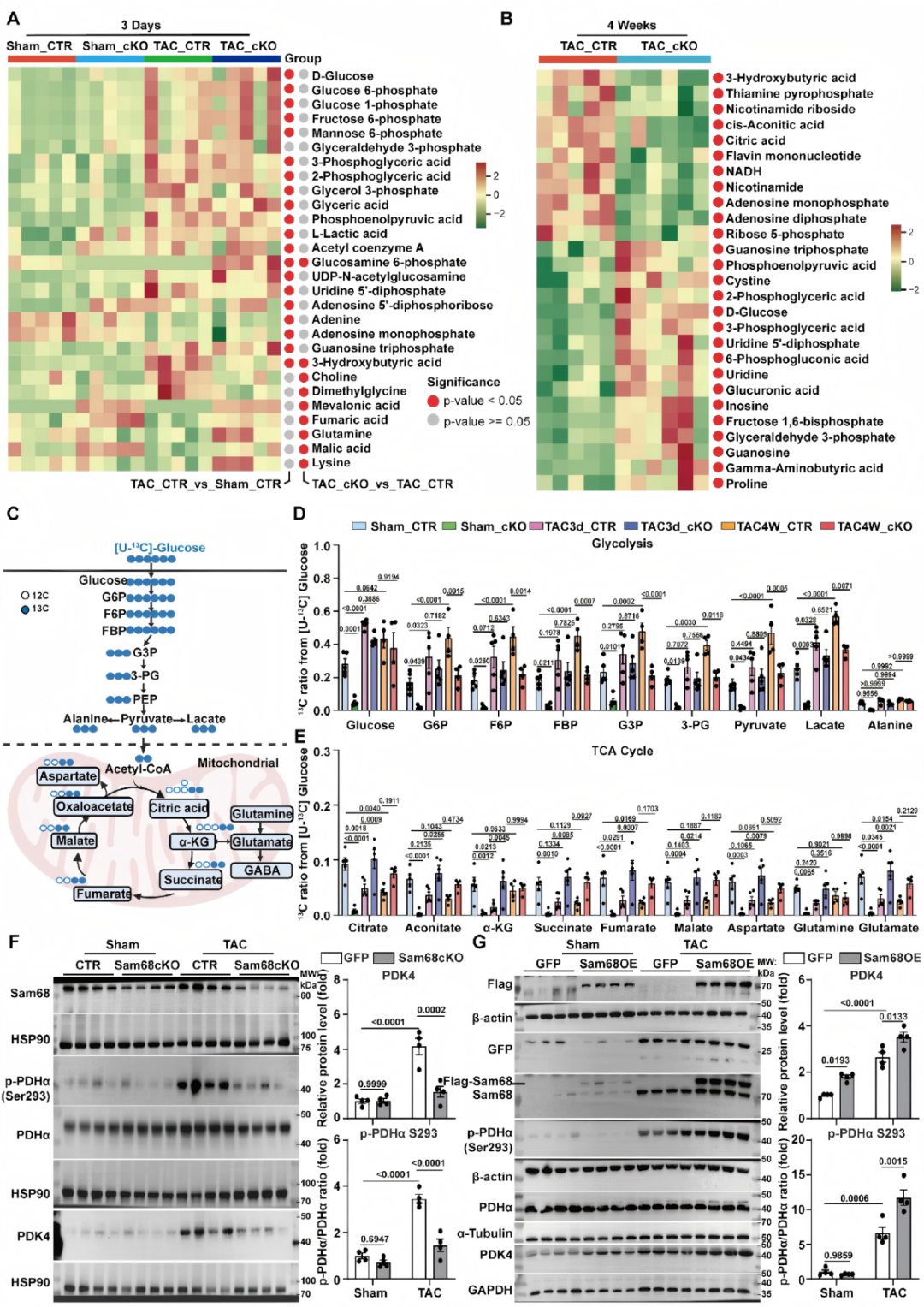
图4.心肌细胞Sam68缺失促进压力超负荷下丙酮酸氧化
05.选择性PDK4抑制剂可逆转Sam68驱动的病理性心肌重构
为验证Sam68是否通过PDK4依赖性PDH抑制加重心肌肥厚,研究者对心肌细胞特异性Sam68过表达(Sam68OE)和对照(GFP)小鼠在TAC术后给予PDK4选择性抑制剂PDK4-IN。结果显示,PDK4-IN显著减轻了TAC诱导的心脏增大、心重指数升高、间质纤维化和心肌细胞肥大,并有效遏制了Sam68OE所致的左心室扩张与收缩功能进一步恶化。分子层面,PDK4-IN降低了PDHα Ser293位点磷酸化和ANP表达,恢复了PDH活性及氧化磷酸化复合物的蛋白水平,但不影响Sam68或PDK4的蛋白丰度。上述结果证实,PDK4介导的PDH抑制是Sam68驱动病理性心肌重构的关键下游效应机制,选择性抑制PDK4可有效拮抗压力超负荷下Sam68促重构作用(图5)。
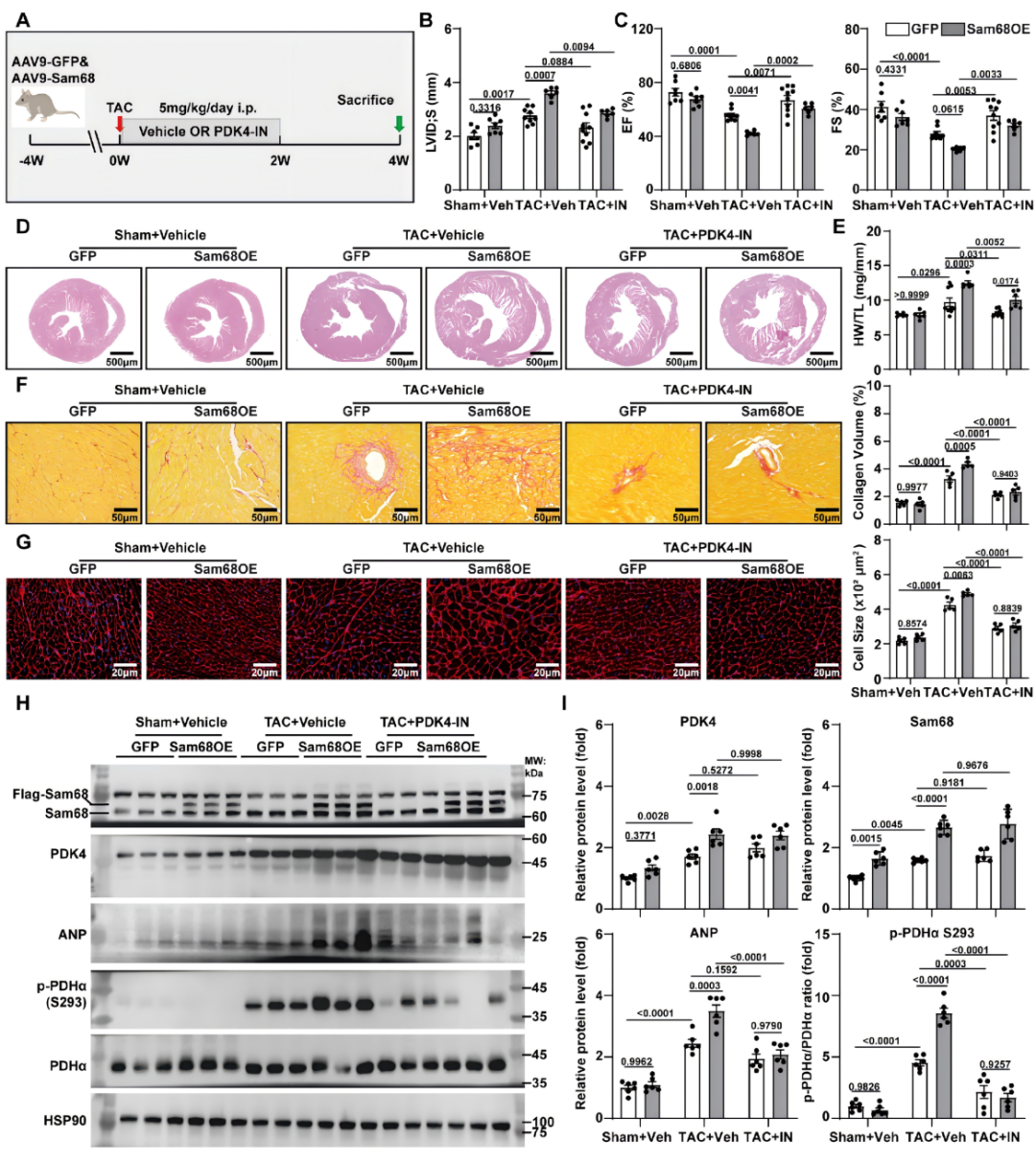
图5.选择性抑制PDK4可逆转Sam68过表达驱动的心肌重构
06.Sam68作为分子支架桥接Src-STAT3信号以驱动PDK4表达
为寻找Sam68调控PDK4的分子机制,研究者首先通过蛋白质相互作用网络分析和分子对接,发现转录因子STAT3可与Sam68直接结合,并在心肌细胞和Sam68过表达心脏中通过免疫共沉淀实验得到验证。功能上,TAC诱导的STAT3 Tyr705磷酸化和核转位在Sam68敲除心脏中被显著抑制,而Sam68过表达则增强其活化;STAT3抑制剂C188-9可完全阻断Sam68依赖的PDK4上调和PDHα磷酸化,证实STAT3是Sam68-PDK4/PDH轴的关键中介。进一步,研究者利用Sam68是Src激酶支架蛋白的特性,证明Sam68通过偶联Src与STAT3形成信号复合物;敲低Sam68或抑制Src均可阻断STAT3 Tyr705磷酸化,且二者无叠加效应;而拟肽抑制剂YB-0158破坏Sam68-Src结合后,可阻止Src-STAT3复合物形成并抑制STAT3核内活化。综上,Sam68在肥厚应激下充当分子支架,偶联Src-STAT3信号模块,从而驱动PDK4转录、抑制PDH活性并损害丙酮酸氧化(图6)。
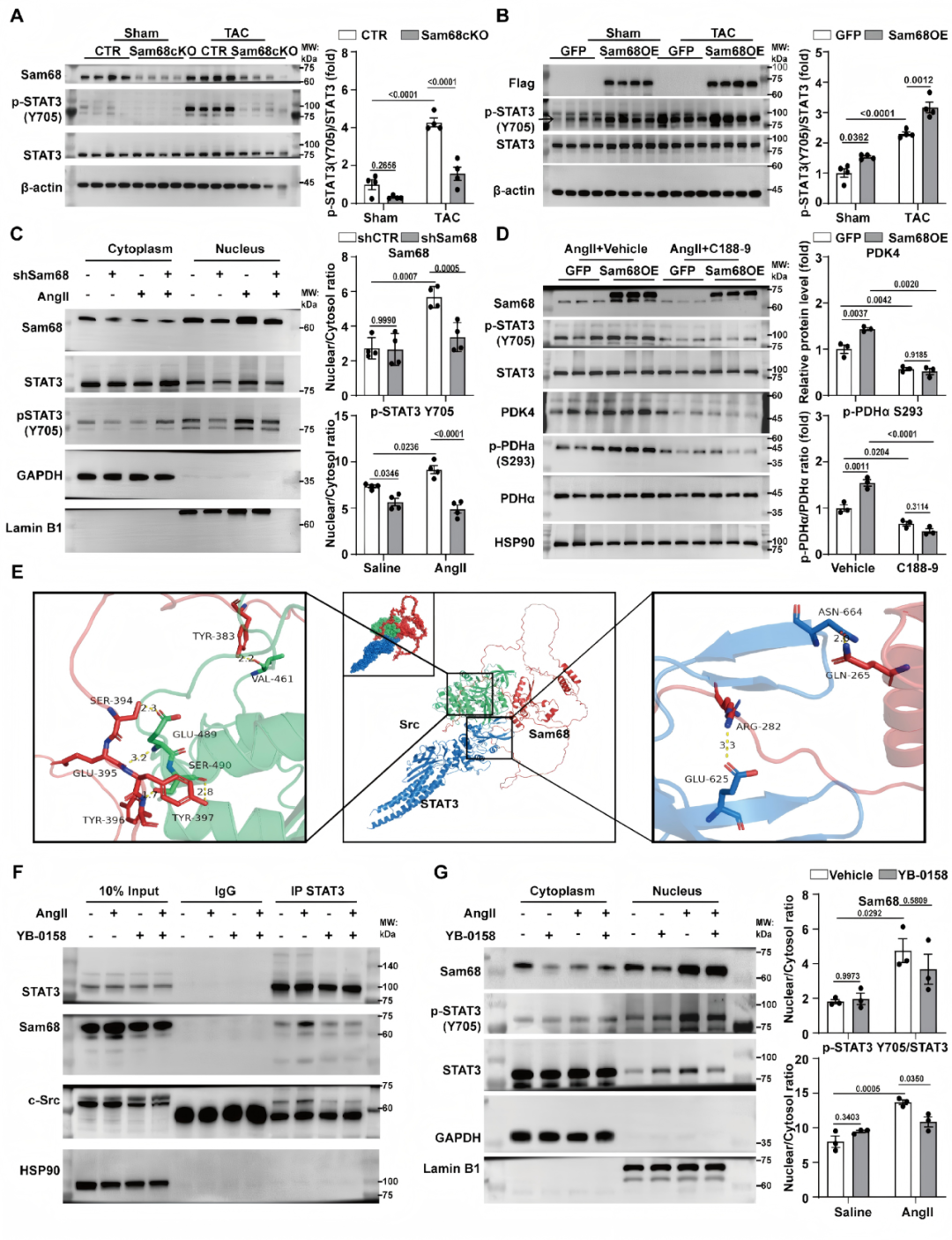
图6.Sam68偶联Src-STAT3信号,驱动STAT3 Tyr705磷酸化及PDK4表达
07.YB-0158通过抑制Sam68减轻TAC诱导的心功能障碍
小分子化合物YB-0158可有效阻断Sam68与Src的相互作用,单次给药即能在心脏中持续抑制Sam68-STAT3复合物形成及STAT3活化。在TAC小鼠模型中,YB-0158显著减轻了心脏重构和功能障碍,其保护作用在Sam68敲除小鼠中几乎消失,证明该药主要通过靶向Sam68发挥心脏保护作用(图7)。
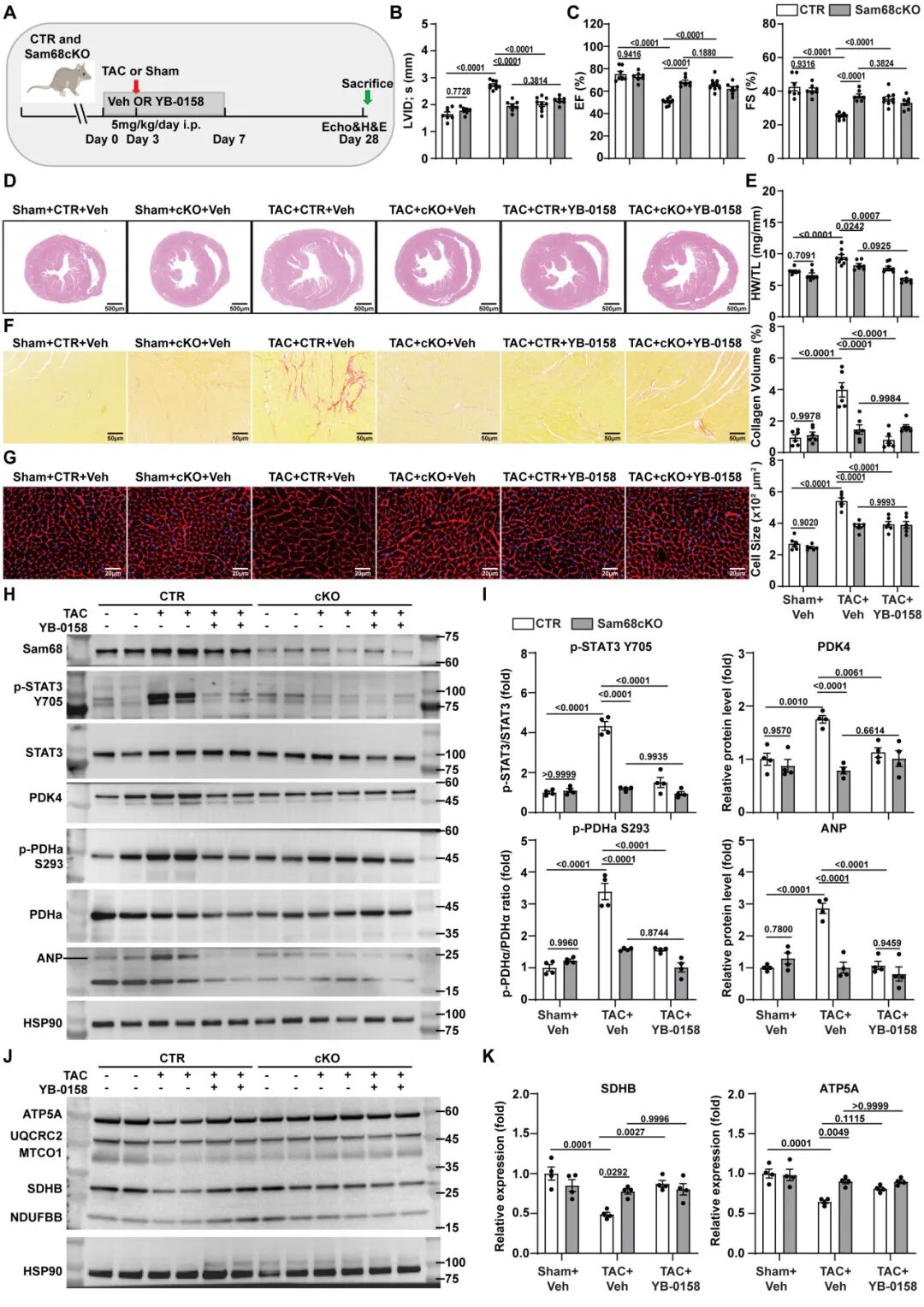
图7.YB-0158通过抑制Sam68减轻病理性心肌肥厚
08.Src/Sam68/STAT3/PDK4信号轴在人类衰竭心脏中被激活
为探究Sam68介导的STAT3-PDK4通路与人类心力衰竭的相关性,研究人员检测了心衰患者与非衰竭对照左心室组织中相关蛋白的表达。结果显示,与对照相比,衰竭心脏中p-Src、Sam68、p-STAT3、PDK4和p-PDH水平均显著升高。总体分析中Sam68蛋白水平与PDK4、ANP及左室射血分数相关,但该关联主要由疾病状态驱动。结合前述实验数据,人类样本结果支持Sam68在衰竭心脏中作为上游调控节点,激活病理性Src-STAT3-PDK4信号轴(图8)。
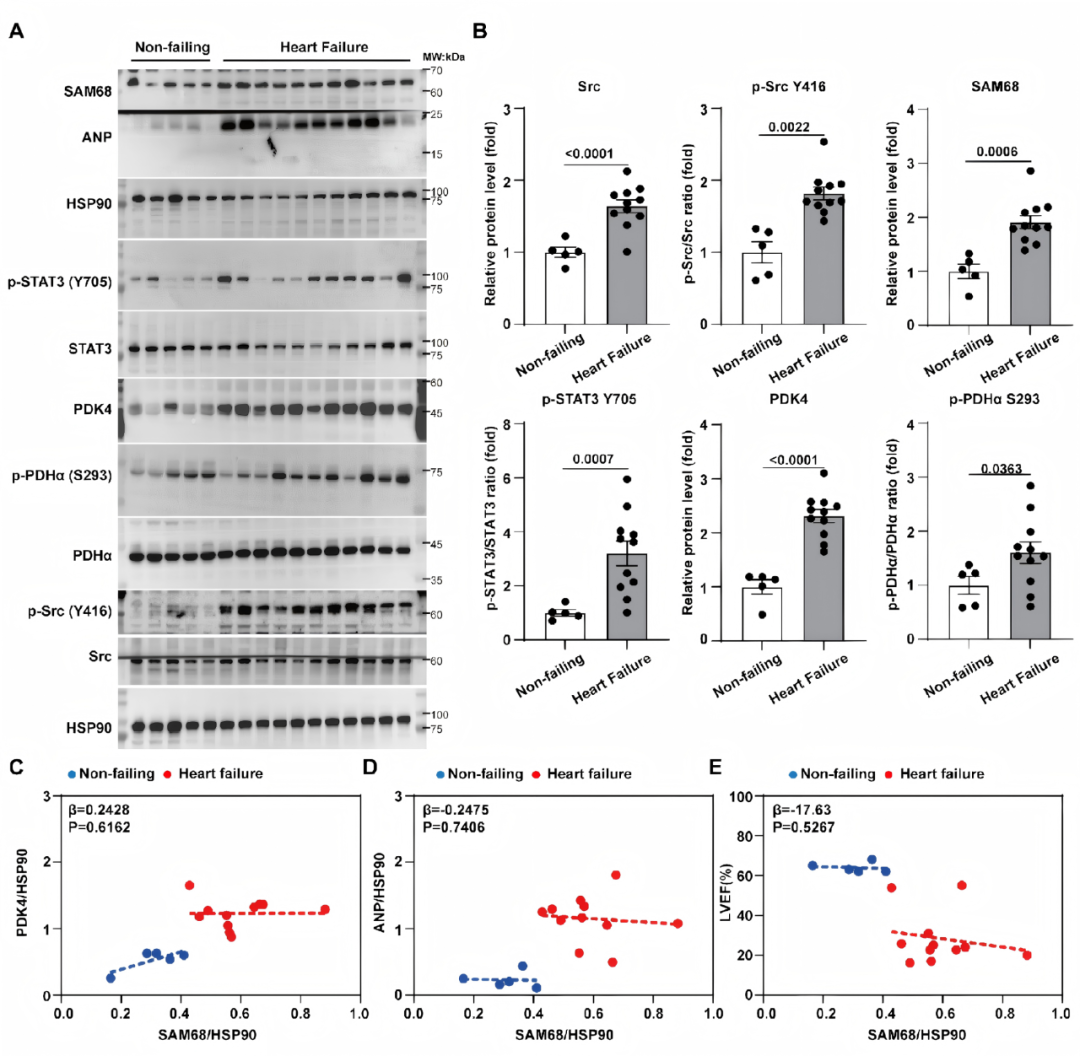
图8.Src/Sam68/STAT3/PDK4信号轴在人类衰竭心脏中被激活
研究总结
综上所述,本研究首次系统阐明了RNA结合蛋白Sam68在病理性心肌肥厚中的关键代谢调控功能。研究揭示了一条全新的"Sam68-Src-STAT3-PDK4"信号轴:在压力超负荷应激下,Sam68作为分子支架偶联Src与STAT3,驱动STAT3 Tyr705磷酸化及核转位,进而上调PDK4表达,导致PDH磷酸化失活、葡萄糖氧化受阻,最终推动心肌代谢失代偿和心衰进展。更重要的是,通过遗传敲除Sam68、选择性抑制PDK4或使用小分子化合物YB-0158阻断Sam68-Src相互作用,均可有效恢复心肌葡萄糖氧化、改善能量代谢和心脏功能。这一发现不仅为理解心力衰竭的代谢发病机制提供了全新视角,更为开发以Sam68为靶点的代谢干预策略奠定了理论基础,有望开启心衰精准治疗的新方向。